To contribute to this effort, we have developed a high-throughput assay for PBPs that measures the fluorescence polarization of Bocillin-FL and used it to screen for potential inhibitors of N. gonorrhoeae PBP 2 from a library of 50,080 compounds. After eliminating those that acted promiscuously, did not demonstrate a concentration-dependent inhibition, or were b-lactams, 18 compounds were examined in more detail, and 7 of these exhibited antimicrobial activity against N. gonorrhoeae, including PenR and CephI strains. To the best of our knowledge, this is the first report of high-throughput screening against a PBP. Interestingly, this compound contains a thiazolidine ring, a feature of penicillin antibiotics and arylalkylidene iminothiazolidin-4-ones, which inhibit some PBPs in vitro and shows antimicrobial activity. Compound 7 was also successful in the docking simulations and there are predicted interactions with Ser310. The resemblance of compound 7 to a b-lactam-like compound led us to determine whether there is a covalent interaction with PBP 2, but no increase in mass of the protein was observed by mass spectrometry. Finally, compound 3 is probably the least promising candidate at this stage since it has a relatively high IC50 against PBP 2 and its MIC values against the N. gonorrhoeae strains are correspondingly weak. Overall, there was relatively weak correlation between IC50 and MIC values for the seven compounds, but this is expected for initial hits from HTS because there are a variety of factors that determine the MIC. Since N. gonorrhoeae expresses two essential PBPs, PBP 1 could be the lethal target for a given compound in addition to, or independent of, PBP 2. The outer membrane of Gram-negative bacteria is a SP600125 molecular weight barrier to hydrophobic compounds and the lipooligosaccharide structure of N. gonorrhoeae also impacts permeability. In addition, entry of hydrophilic compounds into the periplasmis influenced by porins and the action of efflux systems can remove compounds from the periplasm. Both of the antibiotic resistant strains contain a mutation in the promoter of mtrCDE that increases expression of the MtrC-MtrD-MtrE effluxpump. The development of this assay paves the way to screen using larger compound libraries and against variants of PBP 2 that contribute to third-generation cephalosporin resistance. They catalyze the hydrolysis of b- glycosidic bonds between the N-acetylmuramic acid and Nacetylglucosamine repeating units composing the backbone of peptidoglycan, the major constituent of bacterial cell walls. Lysozyme is a component of both phagocytic and secretory granules of neutrophils and is also produced by monocytes, macrophages and epithelial cells. It is found in significant concentrations in saliva, airway mucus, milk and other secretions, and is considered to be an important first line barrier against bacterial infection. While many gram-positive bacteria are rapidly killed by lysozyme in vitro, gram-negative bacteria are not because they have an outer membrane that prevents direct access of lysozyme to the peptidoglycan sacculus. However, in vivo, gramnegative bacteria are sensitized to lysozyme by accessory antimicrobial peptides of the 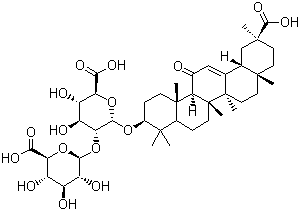 innate immunity system such as defensins and complement which disrupt the outer membrane barrier. Several structurally different lysozymes have been described and the major types within the animal kingdom are the c-type, the R428 g-type and the i-type lysozymes. Vertebrates have genes for both c- and g-type lysozyme, but their spatio-temporal expression is species-specific. The chicken genome for instance comprises a single c-type and two g-type lysozyme genes.
innate immunity system such as defensins and complement which disrupt the outer membrane barrier. Several structurally different lysozymes have been described and the major types within the animal kingdom are the c-type, the R428 g-type and the i-type lysozymes. Vertebrates have genes for both c- and g-type lysozyme, but their spatio-temporal expression is species-specific. The chicken genome for instance comprises a single c-type and two g-type lysozyme genes.
The gene is highly expressed in the oviduct under control of steroid hormones as well as in macrophages
Leave a reply
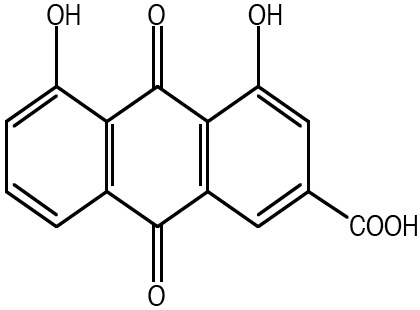 the combination of the N-glycans and the NH2-terminus of PCI affect the rate of its inhibition. Estrogens are important steroidal hormones which exert different physiological functions. The main beneficial effects include their role in programming the breast and uterus for sexual reproduction, controlling cholesterol production in ways that limit the build-up of plaque in the coronary arteries, and preserving bone strength by helping to maintain the proper balance between bone build-up and breakdown. Among female sex hormones, 17b-estradiol is the most potent estrogen carrying out its action either via transactivation of estrogen receptors or by stimulating nongenomic effects via the MAPK signaling pathway. In addition to its important beneficial effects, however, E2 can also cause serious problems arising from its ability to promote the cell proliferation in breast and uterus. Although this is one of the normal functions of estrogen in the body, it can also increase the risk of estrogen dependent diseases, like breast cancer, endometriosis and endometrial hyperplasia. Suppression of estrogenic effects is consequently a major therapeutic approach.
the combination of the N-glycans and the NH2-terminus of PCI affect the rate of its inhibition. Estrogens are important steroidal hormones which exert different physiological functions. The main beneficial effects include their role in programming the breast and uterus for sexual reproduction, controlling cholesterol production in ways that limit the build-up of plaque in the coronary arteries, and preserving bone strength by helping to maintain the proper balance between bone build-up and breakdown. Among female sex hormones, 17b-estradiol is the most potent estrogen carrying out its action either via transactivation of estrogen receptors or by stimulating nongenomic effects via the MAPK signaling pathway. In addition to its important beneficial effects, however, E2 can also cause serious problems arising from its ability to promote the cell proliferation in breast and uterus. Although this is one of the normal functions of estrogen in the body, it can also increase the risk of estrogen dependent diseases, like breast cancer, endometriosis and endometrial hyperplasia. Suppression of estrogenic effects is consequently a major therapeutic approach.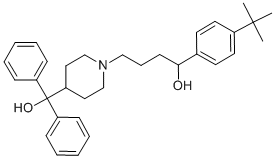 have been categorizing novel ligands for their properties in T-cell differentiation. The above data support that SU5416 enhances Treg generation in vitro, and that IDO is generated in pDCs in response to SU5416 in vitro in an AHR-dependent manner. We continue to characterize these effects for multiple ligands, and are considering theories explaining these differences including the potency and duration of binding of the ligands to the receptor, a possible change in conformation of the receptor when different ligands bind, and a possible effect on APC-T cell interactions. That being said, there is some data to suggest that these dichotomous findings are not as clear cut as originally thought. Most of the in vitro studies examining effects on T-cell
have been categorizing novel ligands for their properties in T-cell differentiation. The above data support that SU5416 enhances Treg generation in vitro, and that IDO is generated in pDCs in response to SU5416 in vitro in an AHR-dependent manner. We continue to characterize these effects for multiple ligands, and are considering theories explaining these differences including the potency and duration of binding of the ligands to the receptor, a possible change in conformation of the receptor when different ligands bind, and a possible effect on APC-T cell interactions. That being said, there is some data to suggest that these dichotomous findings are not as clear cut as originally thought. Most of the in vitro studies examining effects on T-cell 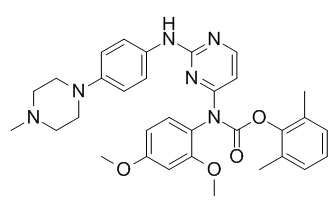 inhibitory small molecules, which are not yet available. Annexin A2 is strongly expressed in lungs, aorta, heart, adrenals and small intestine. Intracellular AnxA2 is part of a heterotetramer complex comprising two AnxA2 monomers and two copies of its natural binding partner, p11. AnxA2 is composed of an N-terminal 23 aa segment that binds p11, followed by four repeat structures. Although lacking a signal peptide, AnxA2 is translocated to the cell surface through a p11-dependent mechanism and is found at the cell surface of epithelial and endothelial cells. As a co-receptor for plasminogen and tissue plasminogen activator, which are required for plasmin generation, the AnxA2 complex promotes vascular fibrinolysis. It was suggested that injection of AnxA2 in human may improve thrombotic disease outcome. AnxA2 knockout mice are viable, but deficient in endothelial plasminogen processing into plasmin and neoangiogenesis. We previously identified AnxA2 as a natural inhibitor of PCSK9’s function on the LDLR, through binding of the first R1 repeat domain of AnxA2 to the CHRD. Inhibition likely occurs via an allosteric PCSK9 conformational change induced by AnxA2, similar to what was reported for mAbs that bind the CHRD. In this report, we investigated in more details the molecular interaction of the R1 domain of AnxA2 and PCSK9 and showed that a synthetic peptide spanning the R1 domain is a potent inhibitor. We further hypothesized that PCSK9 is much more active in enhancing LDLR degradation in AnxA22/2 mice. Indeed, our data showed that AnxA22/2 mice exhibit a higher level of
inhibitory small molecules, which are not yet available. Annexin A2 is strongly expressed in lungs, aorta, heart, adrenals and small intestine. Intracellular AnxA2 is part of a heterotetramer complex comprising two AnxA2 monomers and two copies of its natural binding partner, p11. AnxA2 is composed of an N-terminal 23 aa segment that binds p11, followed by four repeat structures. Although lacking a signal peptide, AnxA2 is translocated to the cell surface through a p11-dependent mechanism and is found at the cell surface of epithelial and endothelial cells. As a co-receptor for plasminogen and tissue plasminogen activator, which are required for plasmin generation, the AnxA2 complex promotes vascular fibrinolysis. It was suggested that injection of AnxA2 in human may improve thrombotic disease outcome. AnxA2 knockout mice are viable, but deficient in endothelial plasminogen processing into plasmin and neoangiogenesis. We previously identified AnxA2 as a natural inhibitor of PCSK9’s function on the LDLR, through binding of the first R1 repeat domain of AnxA2 to the CHRD. Inhibition likely occurs via an allosteric PCSK9 conformational change induced by AnxA2, similar to what was reported for mAbs that bind the CHRD. In this report, we investigated in more details the molecular interaction of the R1 domain of AnxA2 and PCSK9 and showed that a synthetic peptide spanning the R1 domain is a potent inhibitor. We further hypothesized that PCSK9 is much more active in enhancing LDLR degradation in AnxA22/2 mice. Indeed, our data showed that AnxA22/2 mice exhibit a higher level of  pharmacokinetics and pharmacodynamics of different DPP-4 inhibitors, in the settings of CRF, in order to determine the properties of DPP-4 inhibitors to be used in patients with impaired renal function, and investigated the effects of linagliptin on biomarkers of cardiac and renal fibrosis. The results showed that DPP-4 inhibition increases plasma GLP-1 levels, particularly in uremia, suggesting that linagliptin may offer a unique approach for treating uremic cardiomyopathy in CKD patients. The overall goal of the present study was to compare the pharmacokinetic properties of available DPP-4 inhibitors in a rat model of uremic heart disease and select the optimal compound based on these data for the first pharmacodynamics analyses of potential efficacy in this rat model. We have shown that renal impairment does not affect the pharmacokinetics of linagliptin, whereas it increases the exposure of sitagliptin and alogliptin. In the present study, only linagliptin was found not to further aggravate pathological changes of glomerular and tubular markers in rats with CRF, suggesting that it is a safe approach to be used in patients with CRF. Consequently, linagliptin was also the compound of choice to investigate further effects on uremic cardiomyopathy. This is of potential clinical impact, since patients with advanced stages of renal impairment are characterized by a high overall cardiac morbidity and mortality.
pharmacokinetics and pharmacodynamics of different DPP-4 inhibitors, in the settings of CRF, in order to determine the properties of DPP-4 inhibitors to be used in patients with impaired renal function, and investigated the effects of linagliptin on biomarkers of cardiac and renal fibrosis. The results showed that DPP-4 inhibition increases plasma GLP-1 levels, particularly in uremia, suggesting that linagliptin may offer a unique approach for treating uremic cardiomyopathy in CKD patients. The overall goal of the present study was to compare the pharmacokinetic properties of available DPP-4 inhibitors in a rat model of uremic heart disease and select the optimal compound based on these data for the first pharmacodynamics analyses of potential efficacy in this rat model. We have shown that renal impairment does not affect the pharmacokinetics of linagliptin, whereas it increases the exposure of sitagliptin and alogliptin. In the present study, only linagliptin was found not to further aggravate pathological changes of glomerular and tubular markers in rats with CRF, suggesting that it is a safe approach to be used in patients with CRF. Consequently, linagliptin was also the compound of choice to investigate further effects on uremic cardiomyopathy. This is of potential clinical impact, since patients with advanced stages of renal impairment are characterized by a high overall cardiac morbidity and mortality.