The BIBW2992 natural QSIs contribute to host defense against bacteria and both natural and synthetic QSIs have been proposed as promising molecules because they may act synergistically with antibiotics to limit bacterial infection. This article reports the identification of novel QSIs such as the natural plant compound hordenine and the synthetic indoline-2carboxamides, and also demonstrates the QSI-activity of the three human sexual hormones that are estrone, estriol and estradiol. QSIs have been identified in many organisms, plants being the most frequently investigated source of QSI compounds and algae the providers of the most potent ones. Our results revealed the QSI potentiality of alkaloids such as hordenine,1248, and 3492. Hordenin is a natural alkaloid of the phenethylamine class exhibiting a widespread occurrence in plants, including those that are used for human and animal consumption. Following injection, hordenine stimulates the release of norepinephrine in mammals hence acting indirectly as an adrenergic drug. In the literature, alkaloid compounds have been less frequently reported as acting as QSI than aromatic or polyaromatic compounds. Indeed, solenopsin A, a venom alkaloid produced by the fire ant Solenopsis invicta, has been shown to inhibit BYL719 1217486-61-7 biofilm formation, pyocyanin and elastase production as well as the expression of QS-regulated genes lasB, rhlI and lasI in P. aeruginosa. Peters and co-workers also demonstrated that brominated tryptamine-based alkaloids from Flustrafoliacea,a sea bryozoan, inhibit AHL-regulated gene expression using biosensors P. putida, P. putida and E. coli lasR, cepR and luxR coupled to the promoter of lasB, cepI and luxI, respectively. In this study, the QSI activity of human hormones was supported by complementary features. The pure hormones, especially estriol and estrone, affected expression of the QSregulated reporter fusion traG-lacZ and QS-dependent horizontal transfer of the virulence Ti-plasmid in A. tumefaciens. They also decreased the expression of six QS-regulated genes lasI, lasR, lasB, rhlI, rhlR, and rhlA in P. aeruginosa, but none decreased expression of the QS-independent gene aceA. Because of the effect on lasI and rhlI, the AHL concentration was also affected in the presence 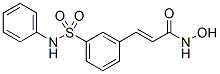 of the sexual hormones. In agreement with a previous report comparing the effect of steroid hormones on the growth of several pathogens, they did not affect the growth of A. tumefaciens and P. aeruginosa at the concentrations used for describing QSI activity. The sexual hormones act as QSIs at a mM-range concentration which is similar to that of the natural polycyclic QSIs such as catechin and naringenin, but higher than that of some other natural and synthetic QSIs which act at a mM range or lower. Our work also revealed that pure hormones affected the QS-regulated reporter gene of P. aeruginosa when RhlR or LasR was expressed in E. coli in the presence of the appropriate AHL. Moreover, molecular modeling confirmed the competitive hormone-binding capacity of the two AHL-sensors LasR and TraR, suggesting that the AHL-LuxR sensors are targets of the discovered QSIs. This mechanism of action is frequently encountered among QSIs. Such a putative cross-talk between QS and hormonal signalling was hypothesized in prospective reviews by Rumbaugh and Hughes and Sperandio and in a paper reporting docking-type screening of QSIs, but, to our knowledge, was never experimentally observed in vitro until this report. Finally, the hypothesis rose about QSI-activity of sexual hormones in vivo because the opportunistic pathogen P. aeruginosa is detectable in several tissues and organs of hospitalized patients and healthy women, and can thus come into contact with sexual hormones. A major argument against this hypothesis is that QSI activity of hormones was observed at 2 mM while, in serum, concentrations of hormones such as estradiol reach up to 0.4�C1.6 nM in healthy women and 2�C18 nM during fertilizing protocols. However, the debate remains still unclosed because clinical and environmental Pseudomonas isolates are known for their capacity to import, bind and biodegrade human hormones, including estrogens, via proteins and pathways that are still poorly-characterized. These hormone-modifying capabilities would contribute to underestimate the QSI-efficiency of hormones in our in vitro assay. Adenosine triphosphate has long been recognized for its role in intracellular energy metabolism.
of the sexual hormones. In agreement with a previous report comparing the effect of steroid hormones on the growth of several pathogens, they did not affect the growth of A. tumefaciens and P. aeruginosa at the concentrations used for describing QSI activity. The sexual hormones act as QSIs at a mM-range concentration which is similar to that of the natural polycyclic QSIs such as catechin and naringenin, but higher than that of some other natural and synthetic QSIs which act at a mM range or lower. Our work also revealed that pure hormones affected the QS-regulated reporter gene of P. aeruginosa when RhlR or LasR was expressed in E. coli in the presence of the appropriate AHL. Moreover, molecular modeling confirmed the competitive hormone-binding capacity of the two AHL-sensors LasR and TraR, suggesting that the AHL-LuxR sensors are targets of the discovered QSIs. This mechanism of action is frequently encountered among QSIs. Such a putative cross-talk between QS and hormonal signalling was hypothesized in prospective reviews by Rumbaugh and Hughes and Sperandio and in a paper reporting docking-type screening of QSIs, but, to our knowledge, was never experimentally observed in vitro until this report. Finally, the hypothesis rose about QSI-activity of sexual hormones in vivo because the opportunistic pathogen P. aeruginosa is detectable in several tissues and organs of hospitalized patients and healthy women, and can thus come into contact with sexual hormones. A major argument against this hypothesis is that QSI activity of hormones was observed at 2 mM while, in serum, concentrations of hormones such as estradiol reach up to 0.4�C1.6 nM in healthy women and 2�C18 nM during fertilizing protocols. However, the debate remains still unclosed because clinical and environmental Pseudomonas isolates are known for their capacity to import, bind and biodegrade human hormones, including estrogens, via proteins and pathways that are still poorly-characterized. These hormone-modifying capabilities would contribute to underestimate the QSI-efficiency of hormones in our in vitro assay. Adenosine triphosphate has long been recognized for its role in intracellular energy metabolism.
Approaches such as the synthesis of structural analogues experimental and virtual screening of chemo-libraries
Leave a reply
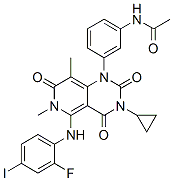 metabolic circuits i.e. the polyol pathway, hexosamine biosynthetic pathway, advanced glycation end products, and PKC activation since previous work found its activation can elicit the onset of cardio-metabolic complications. Since our previous ex vivo rat heart study implicated altered calcium homeostasis in PI-mediated cardiac dysfunction, we further investigated calcium signaling and mitochondrial energetic regulators in an established rat model of chronic PI drug delivery. These data may explain and suggest an association between molecular changes and depressed cardiac contractile function. Although HAART markedly improves the quality of life and prognosis of HIV-infected individuals, it also elicits cardiometabolic side effects in the long-term.
metabolic circuits i.e. the polyol pathway, hexosamine biosynthetic pathway, advanced glycation end products, and PKC activation since previous work found its activation can elicit the onset of cardio-metabolic complications. Since our previous ex vivo rat heart study implicated altered calcium homeostasis in PI-mediated cardiac dysfunction, we further investigated calcium signaling and mitochondrial energetic regulators in an established rat model of chronic PI drug delivery. These data may explain and suggest an association between molecular changes and depressed cardiac contractile function. Although HAART markedly improves the quality of life and prognosis of HIV-infected individuals, it also elicits cardiometabolic side effects in the long-term.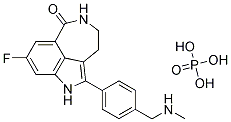 meiotic development, but not vegetative growth. Strikingly, these sporulation-specific inhibitors were structurally related to a class of compounds called cationic amphiphilic drugs, or CADs. Members of this class are weak bases with lipophilic properties, and tend to accumulate in acidic intracellular compartments such as lysosomes. Once inside the acidic milieu of the lysosome, the molecules
meiotic development, but not vegetative growth. Strikingly, these sporulation-specific inhibitors were structurally related to a class of compounds called cationic amphiphilic drugs, or CADs. Members of this class are weak bases with lipophilic properties, and tend to accumulate in acidic intracellular compartments such as lysosomes. Once inside the acidic milieu of the lysosome, the molecules 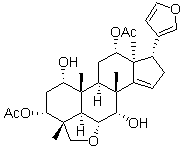 and HDAC inhibitor has been confirmed to be promising in cancer therapeutics. In the current study, we report that LC and Vel combination efficiently exerts anti-tumor effect both in vitro and in vivo. This has been confirmed by the following results. The combinationdecreased cell viability both in hetaptic HepG2 and SMMC-7721 cancer cells;induced cancer cell death in vitro detected by flow cytometry, morphological observation and PARP cleavage;inhibited tumor growth in vivo. Two models for the mechanism of enhancing cytotoxicity by HDAC inhibitors and proteasome inhibitors have been recently proposed. One model is that HDAC inhibitors promote proteasome inhibition-induced proteotoxic stress. By blocking the proteasome, proteasome inhibitors enhance the accumulation of damaged and misfolded proteins, thus inducing downstream free radical accumulation, ER stress and caspase activation; the second is that proteasome inhibitors enhance HDAC inhibition. In this model, HDAC inhibitors serves as the primary cytotoxic stimulus, perhaps by promoting expression of “death genes” via histone acetylation. Based on our findings, two pathways for the crosstalk between HDAC inhibition and proteasome inhibition have been proposed in this study.
and HDAC inhibitor has been confirmed to be promising in cancer therapeutics. In the current study, we report that LC and Vel combination efficiently exerts anti-tumor effect both in vitro and in vivo. This has been confirmed by the following results. The combinationdecreased cell viability both in hetaptic HepG2 and SMMC-7721 cancer cells;induced cancer cell death in vitro detected by flow cytometry, morphological observation and PARP cleavage;inhibited tumor growth in vivo. Two models for the mechanism of enhancing cytotoxicity by HDAC inhibitors and proteasome inhibitors have been recently proposed. One model is that HDAC inhibitors promote proteasome inhibition-induced proteotoxic stress. By blocking the proteasome, proteasome inhibitors enhance the accumulation of damaged and misfolded proteins, thus inducing downstream free radical accumulation, ER stress and caspase activation; the second is that proteasome inhibitors enhance HDAC inhibition. In this model, HDAC inhibitors serves as the primary cytotoxic stimulus, perhaps by promoting expression of “death genes” via histone acetylation. Based on our findings, two pathways for the crosstalk between HDAC inhibition and proteasome inhibition have been proposed in this study. Mtb exposed to an acidic pH, conditions under which Mtb replicates little. Fatty acid synthase-I has been proposed as a target for PZA, but while 5-Clpyrazinamide targets this protein, PZA does not. Recent studies point to RpsA and trans-translation as a target of pyrazinoic acid. It has also been proposed that POA does not have a specific cellular target but simply functions to shuttle protons from the extracellular space into the intrabacterial space, resulting in decreased pHIB, collapse of membrane potential, and bacterial death. Our results provide direct evidence that PZA lowers Mtb��s pHIB in an acidic environment. This assay may select for compounds with similar sterilizing abilities as PZA, an important goal, as resistance to PZA is increasing. We chose to screen a natural product library because of natural products�� structural diversity and greater propensity for antiinfective activity than seen with compounds produced by conventional combinatorial chemistry. A particular challenge in the chemical biology of Mtb is its thick cell wall comprised largely of mycolic acids and their esters.
Mtb exposed to an acidic pH, conditions under which Mtb replicates little. Fatty acid synthase-I has been proposed as a target for PZA, but while 5-Clpyrazinamide targets this protein, PZA does not. Recent studies point to RpsA and trans-translation as a target of pyrazinoic acid. It has also been proposed that POA does not have a specific cellular target but simply functions to shuttle protons from the extracellular space into the intrabacterial space, resulting in decreased pHIB, collapse of membrane potential, and bacterial death. Our results provide direct evidence that PZA lowers Mtb��s pHIB in an acidic environment. This assay may select for compounds with similar sterilizing abilities as PZA, an important goal, as resistance to PZA is increasing. We chose to screen a natural product library because of natural products�� structural diversity and greater propensity for antiinfective activity than seen with compounds produced by conventional combinatorial chemistry. A particular challenge in the chemical biology of Mtb is its thick cell wall comprised largely of mycolic acids and their esters.