Non-covalent and reversible interactions with FG domains from multiple Nups and diffusion through the NPC, whereas interaction with Nup153 might be critical for mediating irreversible and directional exit from the NPC. Interaction with FG-repeats may be mediated indirectly by nuclear transport receptors, such as Mechlorethamine hydrochloride importin 7,, or transportin 3. After exit from the nuclear basket, both Nup98 and Nup153, which shuttle on and off the NPC and have been shown to interact with chromatin,,, might accompany the PIC to its integration site. In particular, Nup98 has been found to localise to the nucleoplasm and participate in chromosomal remodelling and regulation of gene expression. It will be interesting to determine whether Nup98 and/or Nup153 are hijacked by HIV-1 for transport to euchromatin and contribute to specific site selection in expressed genes, since previous work has shown that HIV-1 integrates preferentially within actively transcribed genes. Surprisingly, Nup98 depletion affected HIV-1 and MLV infection equally, but the reduction of MLV infectivity could merely be due to the slight accumulation of cells in G1/S phase that we observed since MLV enters the nucleus during metaphase. Our work demonstrates that a key to the ability of HIV-1 to replicate in non-dividing cells is its capacity to use NPC components for its active transport across the nuclear pore, thus underlining the evolutionary adaptability of HIV-1 to exploit host mechanisms to achieve active nuclear import. Our study suggests a new appealing role for the NPC in HIV-1 infection proposing that the viral nuclear entry step may be important not only for actual translocation, but also for correct subsequent integration as a result of the physical interaction that exists between nuclear pore baskets and the chromatin. The study of the physical and functional interactions between HIV-1 and the NPC not only contributes to our understanding of how other viruses manipulate the nuclear pore but also strengthen our comprehension of lentiviral vectors used for gene transfer protocols, whose active nuclear import is similar to 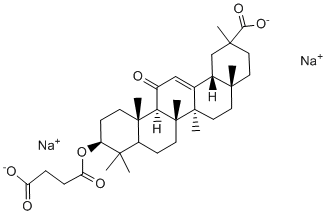 that of HIV-1. From a seed no bigger than that of a cucumber, California’s coastal redwood tree can grow to a height of more than 350 feet. At the same time, aquatic watermeal plants are so small that they resemble specks of cornmeal. The nature of the mechanisms controlling the size and shape of organs is an important but still unanswered question of developmental biology. Two factors determine the size of mature organs: cell number and cell size. A current model for metazoans proposes that cell number and cell size are controlled by distinct proliferation and growth signals that negatively affect each other; when cellular size is increased, cell count is reduced and vice versa. This regulation ensures that induced alterations in cell proliferation are compensated for by changes in cell size, resulting in little net Benzethonium Chloride change in the final organ size. The compensation phenomenon has also been observed in plants �C; however, it is not clear whether the model proposed for metazoans can be directly applied to plants;. First of all, the majority of cell elongation occurs after termination of cell division and, therefore, the actions of proliferation factors and cell expansion factors are separated in time. In addition, some signals, such as auxin, regulate both proliferation and elongation of cells . To understand regulation of organ size and the molecular mechanism of compensation it is essential to know the signaling pathways that control and coordinate cell proliferation and cell elongation. Plant hormones are the most obvious candidates for that role, with cytokinins being implicated in regulation of cell proliferation; and gibberellins, brassinosteroids, and auxin regulating both cell proliferation and cell elongation.
that of HIV-1. From a seed no bigger than that of a cucumber, California’s coastal redwood tree can grow to a height of more than 350 feet. At the same time, aquatic watermeal plants are so small that they resemble specks of cornmeal. The nature of the mechanisms controlling the size and shape of organs is an important but still unanswered question of developmental biology. Two factors determine the size of mature organs: cell number and cell size. A current model for metazoans proposes that cell number and cell size are controlled by distinct proliferation and growth signals that negatively affect each other; when cellular size is increased, cell count is reduced and vice versa. This regulation ensures that induced alterations in cell proliferation are compensated for by changes in cell size, resulting in little net Benzethonium Chloride change in the final organ size. The compensation phenomenon has also been observed in plants �C; however, it is not clear whether the model proposed for metazoans can be directly applied to plants;. First of all, the majority of cell elongation occurs after termination of cell division and, therefore, the actions of proliferation factors and cell expansion factors are separated in time. In addition, some signals, such as auxin, regulate both proliferation and elongation of cells . To understand regulation of organ size and the molecular mechanism of compensation it is essential to know the signaling pathways that control and coordinate cell proliferation and cell elongation. Plant hormones are the most obvious candidates for that role, with cytokinins being implicated in regulation of cell proliferation; and gibberellins, brassinosteroids, and auxin regulating both cell proliferation and cell elongation.
Despite these advances a detailed understanding of the role of hormones involve a series of low-affinity
Leave a reply
 this region is important for normal cardiac development. How can we explain these divergent observations? One possibility is that the SRJ is indeed dispensable for mammalian heart development, and that the observations from patients may be misleading. Supporting this idea, there is considerable lack of conservation in this domain between placental and non-placental mammals. Furthermore, the mutations in the SRJ do not significantly affect the disordered nature of the domain, indicating that it may be able to accommodate mutations without adversely affecting the overall structure and function of the protein. Moreover, in no case has it been shown that the mutation has either arisen de novo or been transmitted from an affected parent. Another possibility that partially reconciles the mouse and human observations is that variants found clustered in the SRJ in CHD patients may not, by themselves.
this region is important for normal cardiac development. How can we explain these divergent observations? One possibility is that the SRJ is indeed dispensable for mammalian heart development, and that the observations from patients may be misleading. Supporting this idea, there is considerable lack of conservation in this domain between placental and non-placental mammals. Furthermore, the mutations in the SRJ do not significantly affect the disordered nature of the domain, indicating that it may be able to accommodate mutations without adversely affecting the overall structure and function of the protein. Moreover, in no case has it been shown that the mutation has either arisen de novo or been transmitted from an affected parent. Another possibility that partially reconciles the mouse and human observations is that variants found clustered in the SRJ in CHD patients may not, by themselves.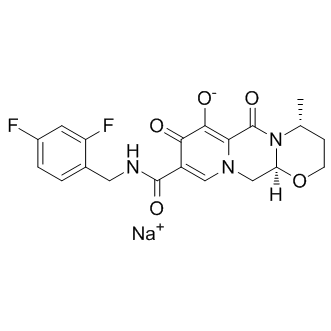 parameters for sensitive and efficient control of a Tet-regulated reporter gene by a protein-based inducer are still largely unknown. While the basic functionality of TIP-mediated induction of TetR has been demonstrated in Escherichia coli and in Staphylococcus aureus, major obstacles must still be overcome, if this system is to be used effectively in more sophisticated applications, like those mentioned above. In the examples published so far, efficient induction of TetR was only achieved after strong overproduction of the TIP-containing fusion protein from a multicopy plasmid.
parameters for sensitive and efficient control of a Tet-regulated reporter gene by a protein-based inducer are still largely unknown. While the basic functionality of TIP-mediated induction of TetR has been demonstrated in Escherichia coli and in Staphylococcus aureus, major obstacles must still be overcome, if this system is to be used effectively in more sophisticated applications, like those mentioned above. In the examples published so far, efficient induction of TetR was only achieved after strong overproduction of the TIP-containing fusion protein from a multicopy plasmid. response or visual discrimination. Thus these findings are consistent with the idea that CIE exposure reduces the ability of the mPFC to flexibly modulate behavior during changing environmental situations that require increased attentional control. Medial PFC networks control executive functions and behavioral flexibility, and in humans they may regulate cognitive control over alcohol intake. Thus the CIEinduced changes in glutamatergic transmission in mPFC pyramidal cells that we describe here may contribute to the cognitive impairments and loss of behavioral control seen in alcoholdependent subjects. On the day following turn bias, mice were trained on a response discrimination task. For this task, the mice were required to always turn in the opposite direction of its turn bias to obtain reward. Mice started from one of 3 arms to discourage them from using an allocentric spatial strategy to locate the food.
response or visual discrimination. Thus these findings are consistent with the idea that CIE exposure reduces the ability of the mPFC to flexibly modulate behavior during changing environmental situations that require increased attentional control. Medial PFC networks control executive functions and behavioral flexibility, and in humans they may regulate cognitive control over alcohol intake. Thus the CIEinduced changes in glutamatergic transmission in mPFC pyramidal cells that we describe here may contribute to the cognitive impairments and loss of behavioral control seen in alcoholdependent subjects. On the day following turn bias, mice were trained on a response discrimination task. For this task, the mice were required to always turn in the opposite direction of its turn bias to obtain reward. Mice started from one of 3 arms to discourage them from using an allocentric spatial strategy to locate the food. In addition, by using a conformation specific antibody anti-Ab oligomers, we demonstrated that hNGFP61S/R100E prevents or reduces the accumulation of Ab oligomers, considered the earliest and most synaptotoxic forms of Ab. Thus, hNGFP61S/R100E exerts an anti-amyloidogenic effect in vivo, in a disease-relevant FAD-based model. From a mechanistic point of view, this might involve a generalized neuroprotective activity by the neurotrophin, at multiple levels, by slowing the generation of Ab peptide and Ab oligomers, by reducing the microgliosis and astrogliosis and/or, possibly, by increasing the clearance of Ab peptides, thereby leading to the observed reduced plaque load and Ab oligomer levels. The observed effects on astrocytes and microglia are consistent with both these cell types expressing TrkA and p75NTR NGF receptors, in normal and pathological conditions. Reactive astrocytes, present in APP xPS1 brain due to amyloid pathology, also express higher levels of TrkA in human AD. We found a reduction of both microgliosis and
In addition, by using a conformation specific antibody anti-Ab oligomers, we demonstrated that hNGFP61S/R100E prevents or reduces the accumulation of Ab oligomers, considered the earliest and most synaptotoxic forms of Ab. Thus, hNGFP61S/R100E exerts an anti-amyloidogenic effect in vivo, in a disease-relevant FAD-based model. From a mechanistic point of view, this might involve a generalized neuroprotective activity by the neurotrophin, at multiple levels, by slowing the generation of Ab peptide and Ab oligomers, by reducing the microgliosis and astrogliosis and/or, possibly, by increasing the clearance of Ab peptides, thereby leading to the observed reduced plaque load and Ab oligomer levels. The observed effects on astrocytes and microglia are consistent with both these cell types expressing TrkA and p75NTR NGF receptors, in normal and pathological conditions. Reactive astrocytes, present in APP xPS1 brain due to amyloid pathology, also express higher levels of TrkA in human AD. We found a reduction of both microgliosis and