This model implies ING1 as a prognostic factor for response to targeted cancer therapy: patients with tumors expressing low ING1-levels might not significantly profit from certain proteasome-inhibitors or other anticancer agents, that involve the induction of p53-dependent apoptosis in the CHIR-99021 252917-06-9 selective killing of tumor cells. Metformin is an oral insulin-sensitizing agent commonly used either alone or in combination with other antihyperglycemic drugs in patients with type 2 diabetes. Based on various population-based analyses, prescription of metformin in patients with type 2 diabetes increased by about 50% in European countries. The glucose-lowering effect of metformin is largely attributable to inhibition of hepatic gluconeogenesis, and additionally, insulinstimulated glucose uptake into skeletal muscle cells and adipocytes is increased by metformin. Recently, it has been shown that organic cation transporters are crucial for the uptake of metformin and these membrane transport proteins are expressed at significant levels in metformin target tissues such as liver, muscle, and adipose tissue. Data from OCT1 knockout mice as well as from healthy volunteers carrying OCT1 variants clearly indicate an alteration of metformin disposition and subsequent consequences for plasma glucose levels. Since metformin does not undergo hepatic metabolism, drug-drug interaction by inhibition of OCT transporters might be important. Because OCT1 is expressed in human liver, alteration of hepatic metformin uptake may be assumed, thereby resulting in poor response to metformin treatment due to reduced glucose-lowering effects. Otherwise, drug-drug interaction with OCT2, which is expressed in proximal tubule epithelial cells, would probably increase systemic disposition of metformin by reduced renal clearance. Recently, a strong inhibiting effect of repaglinide and rosiglitazone on OCT1-mediated metformin transport as well as of several drugs on OCT2-mediated metformin transport in vitro has been reported. Clinically, concomitant use of the potent OCT2 inhibitors cimetidine and verapamil in cisplatin-treated patients resulted in a lower risk for cisplatin-related nephrotoxicity since the antitumor drug cisplatin is an OCT2 substrate. This clinical observation is supported by animal data, clearly demonstrating that cimetidinerelated inhibition of the OCT2 transporter alters cisplatin uptake in the kidney. These examples suggest that OCT-mediated drug-drug interactions appear to be clinically relevant. Hundreds of xenobiotics including drugs potentially inhibiting OCTs were tested in the past and several new inhibitors have been identified. However, systematic data regarding the important drug class of proton 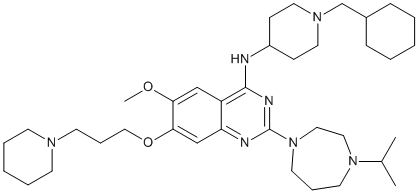 pump inhibitors are still missing although PPIs are frequently used in metformin-treated patients with metabolic syndrome and cardiovascular diseases. Moreover, gastroesophageal reflux disease is commonly seen in patients with type 2 diabetes and PPIs are the drugs of best choice in treatment of GERD. With the recent advances in the understanding of the role of drug transporters in pharmacokinetics it has become critical to elucidate drug-drug interactions that are rooted in transporters. Drug transporters can be generally classified as either uptake or MDV3100 efflux transporters characterizing whether they facilitate drug entry into a cell or efflux out of a cell. In the present paper we focused on the uptake transporter proteins OCT1, OCT2, and OCT3 since the antidiabetic drug metformin is a substrate for each and there is already evidence.
pump inhibitors are still missing although PPIs are frequently used in metformin-treated patients with metabolic syndrome and cardiovascular diseases. Moreover, gastroesophageal reflux disease is commonly seen in patients with type 2 diabetes and PPIs are the drugs of best choice in treatment of GERD. With the recent advances in the understanding of the role of drug transporters in pharmacokinetics it has become critical to elucidate drug-drug interactions that are rooted in transporters. Drug transporters can be generally classified as either uptake or MDV3100 efflux transporters characterizing whether they facilitate drug entry into a cell or efflux out of a cell. In the present paper we focused on the uptake transporter proteins OCT1, OCT2, and OCT3 since the antidiabetic drug metformin is a substrate for each and there is already evidence.
The antidiabetics repaglinide or rosiglitazone as well as H2 receptor antagonists inhibit OCT function
Leave a reply
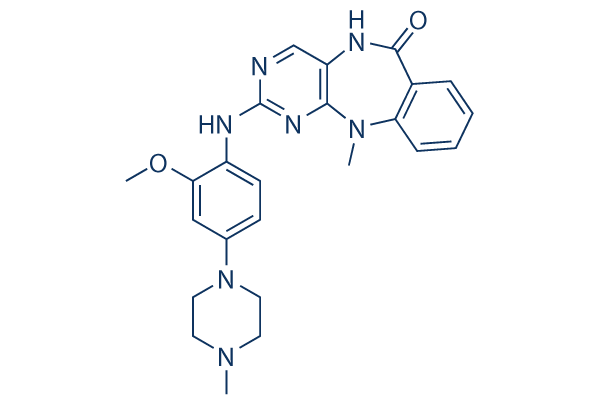 In terms of the differences in Ki values, we speculate that larger substrates may be more capable than smaller ones of effecting the transition between the closed and open configurations, resulting in an increased off rate for the inhibitor. It may also be that the inhibitor can be trapped inside the internal chamber only in the case of smaller substrates. Alternatively, given that 2 residues within Ii1 protrude into the internal chamber, it may be that larger substrates sterically block a subset of binding modes of the inhibitor. In this context, it is relevant to note that larger substrates are known to interact with an exosite present inside the catalytic chamber but opposite to the active site.
In terms of the differences in Ki values, we speculate that larger substrates may be more capable than smaller ones of effecting the transition between the closed and open configurations, resulting in an increased off rate for the inhibitor. It may also be that the inhibitor can be trapped inside the internal chamber only in the case of smaller substrates. Alternatively, given that 2 residues within Ii1 protrude into the internal chamber, it may be that larger substrates sterically block a subset of binding modes of the inhibitor. In this context, it is relevant to note that larger substrates are known to interact with an exosite present inside the catalytic chamber but opposite to the active site.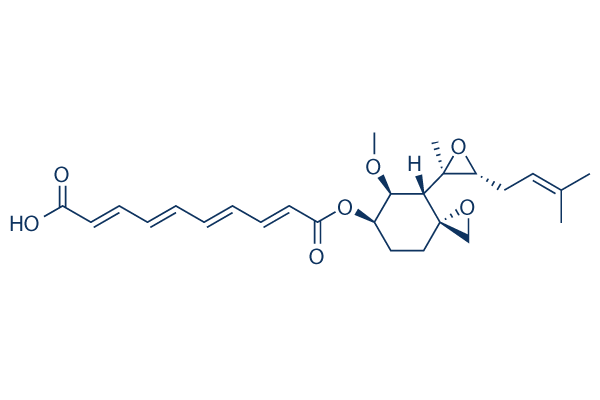 dependent activation of the stress-activated protein kinases p38 MAP kinase and c-Jun N-terminal kinase in HEK-293 cells. During the course of our investigation into the mechanism by which overexpression of TRPM7 caused loss of cell adhesion, we discovered that application of the non-specific lipoxygenase inhibitor NDGA attenuated cell rounding and loss of adhesion produced by influx of divalent cations through the channel. Electrophysiological measurements demonstrated that application of NDGA potently inhibited TRPM7 channel activity, suggesting that a lipoxygenase may be involved in regulating the channel. Lipoxygenases are a family of calciumdependent dioxygenases, including 5-lipoxygenase, 12lipoxygenase, and 15-lipoxygenase, that metabolize arachidonic acid to distinct biologically active fatty acid hydroperoxides, such as leukotrienes and hydroyeicosateraenoic acids. These metabolites take part in numerous cell processes, including inflammation, proliferation, cell invasion, angiogenesis, cell adhesion, and cell spreading. It is well known that lipid molecules modulate several members of the TRP family. In particular, products of lipoxygenase have been demonstrated to directly modulate TRPV1 channel activity. Activation of histamine and bradykinin receptors stimulates TRPV1 channel activity in a 12-LOX-dependent manner. Thus, our
dependent activation of the stress-activated protein kinases p38 MAP kinase and c-Jun N-terminal kinase in HEK-293 cells. During the course of our investigation into the mechanism by which overexpression of TRPM7 caused loss of cell adhesion, we discovered that application of the non-specific lipoxygenase inhibitor NDGA attenuated cell rounding and loss of adhesion produced by influx of divalent cations through the channel. Electrophysiological measurements demonstrated that application of NDGA potently inhibited TRPM7 channel activity, suggesting that a lipoxygenase may be involved in regulating the channel. Lipoxygenases are a family of calciumdependent dioxygenases, including 5-lipoxygenase, 12lipoxygenase, and 15-lipoxygenase, that metabolize arachidonic acid to distinct biologically active fatty acid hydroperoxides, such as leukotrienes and hydroyeicosateraenoic acids. These metabolites take part in numerous cell processes, including inflammation, proliferation, cell invasion, angiogenesis, cell adhesion, and cell spreading. It is well known that lipid molecules modulate several members of the TRP family. In particular, products of lipoxygenase have been demonstrated to directly modulate TRPV1 channel activity. Activation of histamine and bradykinin receptors stimulates TRPV1 channel activity in a 12-LOX-dependent manner. Thus, our 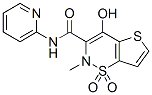 frataxin is incompatible with life in higher organisms, as demonstrated by the embryonic lethality observed in systemic gene knock-out models and by the eventual loss of cells targeted for frataxin gene deletion in conditional knock-out models. The human disease is caused by the pathological hyperexpansion of a GAA?TTC repeat sequence, ranging from 60�C1700 repeats, in the first intron of the frataxin gene that partially suppresses FXN gene expression. This
frataxin is incompatible with life in higher organisms, as demonstrated by the embryonic lethality observed in systemic gene knock-out models and by the eventual loss of cells targeted for frataxin gene deletion in conditional knock-out models. The human disease is caused by the pathological hyperexpansion of a GAA?TTC repeat sequence, ranging from 60�C1700 repeats, in the first intron of the frataxin gene that partially suppresses FXN gene expression. This  innate immunity system such as defensins and complement which disrupt the outer membrane barrier. Several structurally different lysozymes have been described and the major types within the animal kingdom are the c-type, the
innate immunity system such as defensins and complement which disrupt the outer membrane barrier. Several structurally different lysozymes have been described and the major types within the animal kingdom are the c-type, the