In general, it is astonishing how few mutations were observed overall in the 228 patients of the study who have failed therapy. Only 43% of patients had any drug resistance-associated mutation detected. Missing drug pressure due to poor adherence could be a LDN-193189 supply possible explanation for the low prevalence of mutations but it is probably not the major reason because.75% of patients reported to have an excellent adherence. Nevertheless, the prevalence of resistance might be underestimated. Currently used genotypic resistance tests have a population detection limit of only,20%. Additional resistant virus variants might be present at lower levels. The late and rare occurrence of PI/r mutations can be explained by their high genetic barrier compared to NNRTIs. However, the mechanism explaining the lack of resistance to 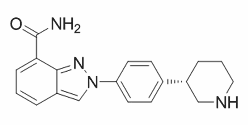 co-administered NRTIs remains unknown. It can be speculated that the two drug classes may have different activities in different anatomical compartments, with regards to free versus cell-cell virus transmissionso that the activity of PI/r might be sufficient to suppress NRTI resistant strains to undetectable levels. It could also be possible that NNRTIs, as they target the same gene as NRTIs, might select for yet unidentified compensatory mutations in the RT connection-, respectively, RNase TWS119 H-domain of the pol gene, subsequently leading to more rapid emergence of NRTI mutations. In theory, the presence of minority variants harboring NNRTI- or NRTI-drug resistant mutations, which have been detected in drug naive HIV-1 infected patients, could have a more severe impact in a regimen that contains a “low genetic barrier” drug rather than a PI/r. This aspect cannot be excluded in the present study.. Poorer adherence in the PI/r-treated group could also possibly explain the differencesbut adherence was excluded as potential bias in a sensitivity analysis. In addition, different NRTI backbones in PI/r- and NNRTI-treated individuals might have influenced our results. To disprove this concern, we performed a sensitivity analysis only including patients with a TDF/FTC backbone and we adjusted the logistic regression for the NRTI backbone. Although our study initially considered 5959 patients who started first-line cART, only 228 individuals qualified for our study. The sample size was too small to compare different treatment regimens in more detail. Unfortunately, sufficient longitudinal resistance data from our patients were not available; otherwise dynamics of evolution of individual drug resistance mutations could have been investigated in more detail. In addition, we cannot exclude that there are resistance associated mutations outside the sequenced region. No phenotypic resistance tests were available that could prove that viruses which do not harbor any mutations are really sensitive to the drugs. In conclusion, PI/r containing cART leads to long-lasting protection of the activity of NRTIs and PI/r despite ongoing viral replication after virological failure. Accumulation of drug resistance mutations against all three drugs of the regimen is slower and less frequent when compared to NNRTI-containing regimens, thus retaining more options for second-line therapy. These findings are of high relevance for settings, which lack the opportunities for regular virological monitoring and where the use of PI/r as first-line therapies should be considered. Inhibiting proteasome function has been demonstrated as a novel therapeutic strategy in multiple disease models like fibrosis, inflammation, ischemia-reperfusion injury and cancer. Proteasome inhibitor bortezomibhas been approved by the United States Food and Drug Administration to treat multiple myeloma.
co-administered NRTIs remains unknown. It can be speculated that the two drug classes may have different activities in different anatomical compartments, with regards to free versus cell-cell virus transmissionso that the activity of PI/r might be sufficient to suppress NRTI resistant strains to undetectable levels. It could also be possible that NNRTIs, as they target the same gene as NRTIs, might select for yet unidentified compensatory mutations in the RT connection-, respectively, RNase TWS119 H-domain of the pol gene, subsequently leading to more rapid emergence of NRTI mutations. In theory, the presence of minority variants harboring NNRTI- or NRTI-drug resistant mutations, which have been detected in drug naive HIV-1 infected patients, could have a more severe impact in a regimen that contains a “low genetic barrier” drug rather than a PI/r. This aspect cannot be excluded in the present study.. Poorer adherence in the PI/r-treated group could also possibly explain the differencesbut adherence was excluded as potential bias in a sensitivity analysis. In addition, different NRTI backbones in PI/r- and NNRTI-treated individuals might have influenced our results. To disprove this concern, we performed a sensitivity analysis only including patients with a TDF/FTC backbone and we adjusted the logistic regression for the NRTI backbone. Although our study initially considered 5959 patients who started first-line cART, only 228 individuals qualified for our study. The sample size was too small to compare different treatment regimens in more detail. Unfortunately, sufficient longitudinal resistance data from our patients were not available; otherwise dynamics of evolution of individual drug resistance mutations could have been investigated in more detail. In addition, we cannot exclude that there are resistance associated mutations outside the sequenced region. No phenotypic resistance tests were available that could prove that viruses which do not harbor any mutations are really sensitive to the drugs. In conclusion, PI/r containing cART leads to long-lasting protection of the activity of NRTIs and PI/r despite ongoing viral replication after virological failure. Accumulation of drug resistance mutations against all three drugs of the regimen is slower and less frequent when compared to NNRTI-containing regimens, thus retaining more options for second-line therapy. These findings are of high relevance for settings, which lack the opportunities for regular virological monitoring and where the use of PI/r as first-line therapies should be considered. Inhibiting proteasome function has been demonstrated as a novel therapeutic strategy in multiple disease models like fibrosis, inflammation, ischemia-reperfusion injury and cancer. Proteasome inhibitor bortezomibhas been approved by the United States Food and Drug Administration to treat multiple myeloma.
Adverse events need also to be taken into account for the choice of first-line treatment
Leave a reply
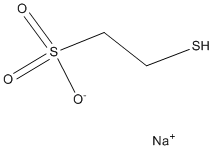 and kill GBM oncospheres. The pattern of effective inhibitor combinations suggests that successful simultaneous inhibition of EGFR and PDGFR and other tyrosine kinases was necessary.
and kill GBM oncospheres. The pattern of effective inhibitor combinations suggests that successful simultaneous inhibition of EGFR and PDGFR and other tyrosine kinases was necessary.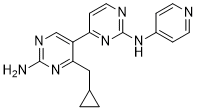 such as DNA repair, cell division, signaling, protein degradation and notably, innate immune function. Ubiquitination occurs by covalent attachment of an,8.5 kDa ubiquitin molecule to a lysine residue in the target protein by the sequential action of three enzymes; a ubiquitin-activating enzyme, a ubiquitin-conjugating enzyme and a ubiquitin-ligase enzyme. Ubiquitin is removed from proteins by deubiquitinases by proteolysis. The human genome encodes over 100 proteins that possess putative DUB activity but physiological substrates of these proteins remain poorly defined for most. DUB enzymes have established roles in a broad spectrum of diseases such as cancer, viral infection and neurodegenerative disorders. Although the function of most DUBs in immune regulation is not known, a few are key players in the modulation of innate immunity and inflammation. For example, the deubiquitinases, A20 and CYLD, control NF-kB signaling, a critical pathway in immunity and cell survival.
such as DNA repair, cell division, signaling, protein degradation and notably, innate immune function. Ubiquitination occurs by covalent attachment of an,8.5 kDa ubiquitin molecule to a lysine residue in the target protein by the sequential action of three enzymes; a ubiquitin-activating enzyme, a ubiquitin-conjugating enzyme and a ubiquitin-ligase enzyme. Ubiquitin is removed from proteins by deubiquitinases by proteolysis. The human genome encodes over 100 proteins that possess putative DUB activity but physiological substrates of these proteins remain poorly defined for most. DUB enzymes have established roles in a broad spectrum of diseases such as cancer, viral infection and neurodegenerative disorders. Although the function of most DUBs in immune regulation is not known, a few are key players in the modulation of innate immunity and inflammation. For example, the deubiquitinases, A20 and CYLD, control NF-kB signaling, a critical pathway in immunity and cell survival.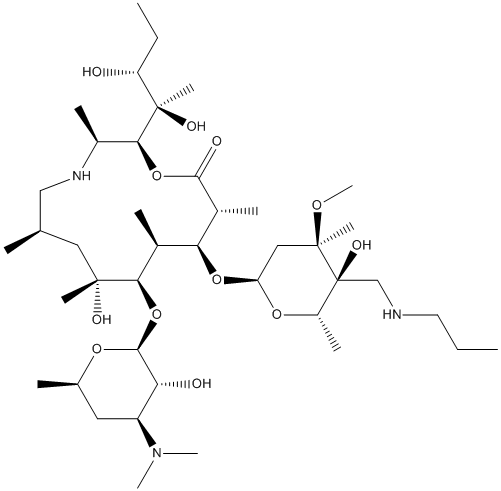 problems, telomeres shorten each time human somatic cells divide and this attrition limits their lifespan. Once the shortest telomere become uncapped, a DNA damage response is induced that mobilizes the p53 and p16/pRB pathways, which then act together to induce senescence, a viable state of irreversible quiescence. If the p53 and p16/pRB pathways are disabled, the cells will ignore these growth inhibitory signals and will continue to divide and shorten their telomeres. Eventually, terminal telomere shortening lead to crisis, a non-viable state associated with programmed cell death. Crisis is triggered by recurrent cycles of telomeretelomere fusions, anaphase bridges and chromosome breakage. When present, telomerase can prevent the induction of senescence and crisis and extend cellular lifespan by the synthesis and addition of new telomeric repeats to the telomeres. Telomerase is ubiquitously present in the early stages of human development. But by the time of birth, expression of the enzyme is repressed and telomerase becomes absent from most somatic tissues, including the pancreas. Cancer specimens, in stark contrast to normal tissues, are
problems, telomeres shorten each time human somatic cells divide and this attrition limits their lifespan. Once the shortest telomere become uncapped, a DNA damage response is induced that mobilizes the p53 and p16/pRB pathways, which then act together to induce senescence, a viable state of irreversible quiescence. If the p53 and p16/pRB pathways are disabled, the cells will ignore these growth inhibitory signals and will continue to divide and shorten their telomeres. Eventually, terminal telomere shortening lead to crisis, a non-viable state associated with programmed cell death. Crisis is triggered by recurrent cycles of telomeretelomere fusions, anaphase bridges and chromosome breakage. When present, telomerase can prevent the induction of senescence and crisis and extend cellular lifespan by the synthesis and addition of new telomeric repeats to the telomeres. Telomerase is ubiquitously present in the early stages of human development. But by the time of birth, expression of the enzyme is repressed and telomerase becomes absent from most somatic tissues, including the pancreas. Cancer specimens, in stark contrast to normal tissues, are