The above two Mechlorethamine hydrochloride methods are predictive approaches that provide with novel, testable small molecule-target associations, while the text mining-based approach is a way to gather information previously existing in the literature that would probably have been missed. Additionally, it also suffers from an inability to detect new 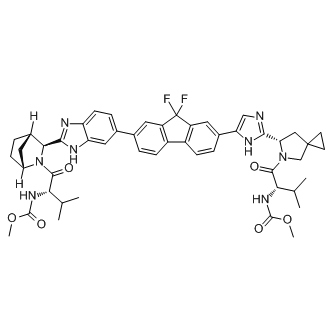 biological findings, and their efficiency is generally hampered by the redundancy of the compound and gene names in literature. Therefore, the genome-wide application of LBVS, SBVS and texting miningbased methods still has many limitations. An effective means that might overcome these problems is not to considerate each drug or target independently from other drugs or targets, but to take the standpoint of chemical genomics which could open up new opportunities to identify new drug leads or therapeutic targets instead. Chemical genomics aims at exploiting the whole chemical space, which corresponds to not only the space of the small molecules but also of those proteins interacting with the molecules. Recently, several chemical genomics approaches, including the ligand-based, targetbased or target-ligand methods have been developed to predict the interactions between compounds and proteins. The ligand-based method that integrated the protein targets was designed at the level of families or subfamilies which is appropriate for some specific protein families such as GPCRs. Based on the ligand binding site similarity, Frimurer et al developed a target-based approach which clustered the Albaspidin-AA receptors and pooled together the known ligands for each cluster to infer shared ligands. Different from these two approaches, the target-ligand approach combines the ligand chemical space, target space and the currently known drug-target networks information to construct a complex forecast system, with purpose to predict ligands or targets for a given target or ligand without prior attempting to define a special set of similar receptors or ligands. For instance, the amino acid sequences, 2-dimensional chemical structures and mass-spectrometry data have been collected together to build a statistical method for predicting compound-protein interactions based on 519 approved drugs and their 291 associated targets. Similarly, the chemical functional groups and biological features have also been adopted to establish the classification models for predicting the drug-target interaction network. Interestingly, without the negative samples, the semi- supervised machine learning algorithm NetLapRLS has been developed based on heterogeneous biological data, which could effectively predict the interaction of each chemical-protein pair. Furthermore, based on DrugBank database, the sets of chemical substructures and protein domains have also been collected and effectively analyzed using Sparse Canonical Correspondence Analysis and Support Vector Machine methods to identify molecular recognition rules between drugs and targets. However, all these aforementioned methods might suffer from the small receptor space which only focused on certain protein families or the limited chemical space only covered by the FDA approved drugs. To predict the drug-target interactions, we have designed a set of in silico tools by incorporating the chemical, genomic and pharmacological information into an integrated framework using the largest available dataset of DrugBank database. The predictions are based on extraction of conserved patterns from subdivided interaction vectors involving both proteins and their corresponding ligands.
biological findings, and their efficiency is generally hampered by the redundancy of the compound and gene names in literature. Therefore, the genome-wide application of LBVS, SBVS and texting miningbased methods still has many limitations. An effective means that might overcome these problems is not to considerate each drug or target independently from other drugs or targets, but to take the standpoint of chemical genomics which could open up new opportunities to identify new drug leads or therapeutic targets instead. Chemical genomics aims at exploiting the whole chemical space, which corresponds to not only the space of the small molecules but also of those proteins interacting with the molecules. Recently, several chemical genomics approaches, including the ligand-based, targetbased or target-ligand methods have been developed to predict the interactions between compounds and proteins. The ligand-based method that integrated the protein targets was designed at the level of families or subfamilies which is appropriate for some specific protein families such as GPCRs. Based on the ligand binding site similarity, Frimurer et al developed a target-based approach which clustered the Albaspidin-AA receptors and pooled together the known ligands for each cluster to infer shared ligands. Different from these two approaches, the target-ligand approach combines the ligand chemical space, target space and the currently known drug-target networks information to construct a complex forecast system, with purpose to predict ligands or targets for a given target or ligand without prior attempting to define a special set of similar receptors or ligands. For instance, the amino acid sequences, 2-dimensional chemical structures and mass-spectrometry data have been collected together to build a statistical method for predicting compound-protein interactions based on 519 approved drugs and their 291 associated targets. Similarly, the chemical functional groups and biological features have also been adopted to establish the classification models for predicting the drug-target interaction network. Interestingly, without the negative samples, the semi- supervised machine learning algorithm NetLapRLS has been developed based on heterogeneous biological data, which could effectively predict the interaction of each chemical-protein pair. Furthermore, based on DrugBank database, the sets of chemical substructures and protein domains have also been collected and effectively analyzed using Sparse Canonical Correspondence Analysis and Support Vector Machine methods to identify molecular recognition rules between drugs and targets. However, all these aforementioned methods might suffer from the small receptor space which only focused on certain protein families or the limited chemical space only covered by the FDA approved drugs. To predict the drug-target interactions, we have designed a set of in silico tools by incorporating the chemical, genomic and pharmacological information into an integrated framework using the largest available dataset of DrugBank database. The predictions are based on extraction of conserved patterns from subdivided interaction vectors involving both proteins and their corresponding ligands.
Allows us to take proteins of different families to the choice of protein encoding by the structural and physicochem
Leave a reply
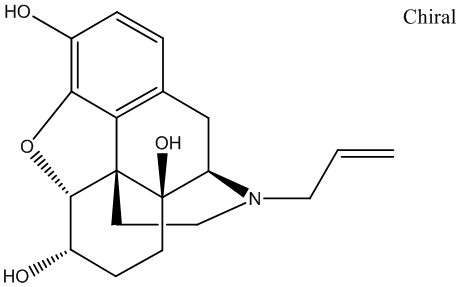 either shift from the previously learned strategy or to maintain the new strategy after perseveration had ceased. In order to detect shifts in the strategies that animals used, trials were separated into consecutive blocks of four trials each. A perseverative error occurred when a mouse made the same egocentric response as required during the Response Discrimination day, but which was opposite to the direction of the arm containing the visual cue. Six of every 12 consecutive trials required the mouse to respond in this manner. A perseverative error was scored when the mouse entered the incorrect arm on three or more trials per block of 4 trials. Once the mouse made less than three perseverative errors in a block, all subsequent errors were now scored as regressive errors. The third type of error, termed “never reinforced” errors, was scored when a mouse entered the incorrect arm on trials where the visual cue was placed on the same side that the mouse had been trained to enter on the previous day. Current strategies for NGF therapy in AD use highly invasive approaches, such as a neurosurgical intracerebroventricular injection of NGF or a parenchimal injection of cells secreting hNGF or of viruses harboring hNGF gene. To fully exploit the therapeutic potential of NGF in a noninvasive manner, its therapeutic window must be improved, by increasing the brain distribution, while limiting NGF paininducing actions. The intranasal delivery represents a viable option to non invasively increase NGF biodistribution in the brain, where it exerts anti-neurodegenerative actions. NGF intranasal delivery minimizes the build-up of peripheral NGF concentration, even if residual leakage and absorption of NGF into the blood stream, from the nasal compartment, has been shown. The fact that the NGF mutation R100W appears to separate the effects of NGF on CNS development from those involved in the activation of peripheral pain pathways, provides a basis for designing “painless” NGF variant molecules. In
either shift from the previously learned strategy or to maintain the new strategy after perseveration had ceased. In order to detect shifts in the strategies that animals used, trials were separated into consecutive blocks of four trials each. A perseverative error occurred when a mouse made the same egocentric response as required during the Response Discrimination day, but which was opposite to the direction of the arm containing the visual cue. Six of every 12 consecutive trials required the mouse to respond in this manner. A perseverative error was scored when the mouse entered the incorrect arm on three or more trials per block of 4 trials. Once the mouse made less than three perseverative errors in a block, all subsequent errors were now scored as regressive errors. The third type of error, termed “never reinforced” errors, was scored when a mouse entered the incorrect arm on trials where the visual cue was placed on the same side that the mouse had been trained to enter on the previous day. Current strategies for NGF therapy in AD use highly invasive approaches, such as a neurosurgical intracerebroventricular injection of NGF or a parenchimal injection of cells secreting hNGF or of viruses harboring hNGF gene. To fully exploit the therapeutic potential of NGF in a noninvasive manner, its therapeutic window must be improved, by increasing the brain distribution, while limiting NGF paininducing actions. The intranasal delivery represents a viable option to non invasively increase NGF biodistribution in the brain, where it exerts anti-neurodegenerative actions. NGF intranasal delivery minimizes the build-up of peripheral NGF concentration, even if residual leakage and absorption of NGF into the blood stream, from the nasal compartment, has been shown. The fact that the NGF mutation R100W appears to separate the effects of NGF on CNS development from those involved in the activation of peripheral pain pathways, provides a basis for designing “painless” NGF variant molecules. In 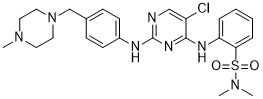 impervious to the action of the Yersinia antiphagocytic machinery. Importantly, virulence effectors were translocated to the same extent into cells regardless of their RACK1 expression level and these intracellular effectors still induced a normal cytotoxic response in both cell lines. This means two things; firstly, translocation of effectors and the process of cytotoxicity induction do not require RACK1, and secondly, antiphagocytosis and cytotoxicity are clearly two distinct events during cell infection. Another striking feature was that the ability of the pathogen to mediate antiphagocytosis towards RACK1 RNAi cells diminished significantly when YopK was present. Notably, this failure to inactivate the critical cellular targets and interrupt phagocytosis occurred despite efficient antiphagocytic Yop effector translocation into the target cells. Hence, the discrepancy between Yersinia YopK+ bacteria inducing an uncompromising cytotoxic response on the one hand, but failing to establish antiphagocytosis on the other, suggests that the latter process requires specific features of the effector translocation process that are overseen by YopK. This agrees with our previous results concerning the antiphagocytic effector and major cytotoxin YopE; certain yopE point mutants exhibited attenuated virulence but still gave rise to cytotoxicity in HeLa cells. Thus, the cytotoxic effect is actually a secondary event that occurs only after prolonged infection and is dispensable for in vivo virulence. On the other hand, there appears to be a direct correlation between the ability of Y. pseudotuberculosis to resist phagocytosis and to cause systemic infections in mice.
impervious to the action of the Yersinia antiphagocytic machinery. Importantly, virulence effectors were translocated to the same extent into cells regardless of their RACK1 expression level and these intracellular effectors still induced a normal cytotoxic response in both cell lines. This means two things; firstly, translocation of effectors and the process of cytotoxicity induction do not require RACK1, and secondly, antiphagocytosis and cytotoxicity are clearly two distinct events during cell infection. Another striking feature was that the ability of the pathogen to mediate antiphagocytosis towards RACK1 RNAi cells diminished significantly when YopK was present. Notably, this failure to inactivate the critical cellular targets and interrupt phagocytosis occurred despite efficient antiphagocytic Yop effector translocation into the target cells. Hence, the discrepancy between Yersinia YopK+ bacteria inducing an uncompromising cytotoxic response on the one hand, but failing to establish antiphagocytosis on the other, suggests that the latter process requires specific features of the effector translocation process that are overseen by YopK. This agrees with our previous results concerning the antiphagocytic effector and major cytotoxin YopE; certain yopE point mutants exhibited attenuated virulence but still gave rise to cytotoxicity in HeLa cells. Thus, the cytotoxic effect is actually a secondary event that occurs only after prolonged infection and is dispensable for in vivo virulence. On the other hand, there appears to be a direct correlation between the ability of Y. pseudotuberculosis to resist phagocytosis and to cause systemic infections in mice.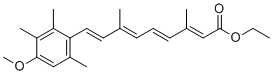 chromatography has been reported in another mouse model for cataracts. Additionally, the mutant mice accumulated aB-crystallinvimentin aggregates in lens cells and aB-crystallin-desmin aggregates in muscle cells. The mechanism of lens opacification likely involves a change in the interaction between vimentin and aB-crystallin. In heterozygous lenses, the interaction between vimentin and aB-crystallin was elevated, even in the absence of significant opacification. This increased interaction and aggregate formation were also observed in the lens epithelial zone of the mutant lenses by immunofluorescence analysis, consistent with published studies using cultured cells. These results indicate that the interaction between the intermediate filament protein vimentin and aB-crystallin may be a precursor to the development of opacities in the mutant lenses. Notably, the mice used in this study were of a mixed 129Sv and C57BL6 background, and the129Sv strain of mice is known to lack a lens-specific intermediate filament known as the beaded filament, although mice lacking the beaded filament do not develop significant opacities and changes in vimentin levels. It would be interesting to determine whether cataract development in aBR120G knock-in mice is altered in mouse strains that have the full complement of the beaded filaments. aB-crystallin is expressed in the cornea where it is important for corneal clarity. Consistent with this, a proportion of the R120G mutant mice developed corneal opacities. However, it is unclear why only a proportion of animals showed this effect. More of the animals are likely to develop corneal opacities as they age, and studies are in progress to assess this possibility. Patients with mutations in the aA-crystallin gene develop similar microcornea and corneal opacities, substantiating our findings in knockin mice and indicating that aB-crystallin also plays a critical role in the maintenance of corneal clarity. A small but significant fraction of the aB-R120G mutant mice had smaller eyes than wild-type littermates, although this small eye phenotype was not as prominent as in homozygous aA-R49C mutant mice. DRMs are a growing class of skeletal muscle disorders caused by mutations in desmin, aB-crystallin, Z-band alternatively spliced PDZ motif, myotilin, filamin C, and Bag3.
chromatography has been reported in another mouse model for cataracts. Additionally, the mutant mice accumulated aB-crystallinvimentin aggregates in lens cells and aB-crystallin-desmin aggregates in muscle cells. The mechanism of lens opacification likely involves a change in the interaction between vimentin and aB-crystallin. In heterozygous lenses, the interaction between vimentin and aB-crystallin was elevated, even in the absence of significant opacification. This increased interaction and aggregate formation were also observed in the lens epithelial zone of the mutant lenses by immunofluorescence analysis, consistent with published studies using cultured cells. These results indicate that the interaction between the intermediate filament protein vimentin and aB-crystallin may be a precursor to the development of opacities in the mutant lenses. Notably, the mice used in this study were of a mixed 129Sv and C57BL6 background, and the129Sv strain of mice is known to lack a lens-specific intermediate filament known as the beaded filament, although mice lacking the beaded filament do not develop significant opacities and changes in vimentin levels. It would be interesting to determine whether cataract development in aBR120G knock-in mice is altered in mouse strains that have the full complement of the beaded filaments. aB-crystallin is expressed in the cornea where it is important for corneal clarity. Consistent with this, a proportion of the R120G mutant mice developed corneal opacities. However, it is unclear why only a proportion of animals showed this effect. More of the animals are likely to develop corneal opacities as they age, and studies are in progress to assess this possibility. Patients with mutations in the aA-crystallin gene develop similar microcornea and corneal opacities, substantiating our findings in knockin mice and indicating that aB-crystallin also plays a critical role in the maintenance of corneal clarity. A small but significant fraction of the aB-R120G mutant mice had smaller eyes than wild-type littermates, although this small eye phenotype was not as prominent as in homozygous aA-R49C mutant mice. DRMs are a growing class of skeletal muscle disorders caused by mutations in desmin, aB-crystallin, Z-band alternatively spliced PDZ motif, myotilin, filamin C, and Bag3.  binds to tubulin b in mesangial cells, supporting our results.
binds to tubulin b in mesangial cells, supporting our results.