Statins have an unparalleled safety and efficacy profile, but often lead to suboptimal levels of LDLc in patients with ADH, show variable patient-dependent responses, and/or result in unwanted side effects, emphasizing the need for other molecules to further lower LDLc. In hepatocytes, statins up-regulate PCSK9 mRNA to a greater extent than LDLR. This revealed the paradox that statins on the one hand enhance LDLR level and activity thereby lowering LDLc, but on the other hand increase the expression of PCSK9 that has the ability to destroy the LDLR and oppose its LDLlowering effect. Therefore, it is believed that neutralization of PCSK9 would enhance the efficacy of statins. Indeed, a significant association of the LOF mutation PCSK9-R46L with statin response was observed in a genome-wide analysis. This supports the hypothesis that the up-regulation of PCSK9 induced by statins attenuates the decrease in LDLc. Lowering PCSK9 levels and/or function has been achieved by antisense mRNA, locked nucleic acids and inhibition of PCSK9;LDLR interaction and degradation using PCSK9 monoclonal antibodies. The latter approach is expensive, restricting it to high risk patients in whom a maximal tolerable dose of statin does not achieve LDLc target levels. Thus, there is a need for cheaper, more accessible 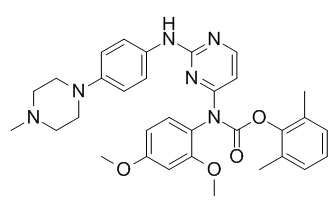 inhibitory small molecules, which are not yet available. Annexin A2 is strongly expressed in lungs, aorta, heart, adrenals and small intestine. Intracellular AnxA2 is part of a heterotetramer complex comprising two AnxA2 monomers and two copies of its natural binding partner, p11. AnxA2 is composed of an N-terminal 23 aa segment that binds p11, followed by four repeat structures. Although lacking a signal peptide, AnxA2 is translocated to the cell surface through a p11-dependent mechanism and is found at the cell surface of epithelial and endothelial cells. As a co-receptor for plasminogen and tissue plasminogen activator, which are required for plasmin generation, the AnxA2 complex promotes vascular fibrinolysis. It was suggested that injection of AnxA2 in human may improve thrombotic disease outcome. AnxA2 knockout mice are viable, but deficient in endothelial plasminogen processing into plasmin and neoangiogenesis. We previously identified AnxA2 as a natural inhibitor of PCSK9’s function on the LDLR, through binding of the first R1 repeat domain of AnxA2 to the CHRD. Inhibition likely occurs via an allosteric PCSK9 conformational change induced by AnxA2, similar to what was reported for mAbs that bind the CHRD. In this report, we investigated in more details the molecular interaction of the R1 domain of AnxA2 and PCSK9 and showed that a synthetic peptide spanning the R1 domain is a potent inhibitor. We further hypothesized that PCSK9 is much more active in enhancing LDLR degradation in AnxA22/2 mice. Indeed, our data showed that AnxA22/2 mice exhibit a higher level of MLN4924 circulating PCSK9 and lower LDLR protein levels in various tissues previously reported to be refractory to PCSK9’s extracellular function on LDLR, such as the adrenals. Furthermore, overexpression of AnxA2 using a recombinant adenovirus resulted in higher LDLR levels in liver. Finally, by sequencing exons of human AnxA2 we identified individuals with a V98L polymorphic variation in the R1 domain, and showed that this variant could be associated with lower levels of circulating PCSK9. The discovery of PCSK9 and its genetic relation to hypercholesterolemia led to a very exciting and active period of identification of the mechanisms of action of PCSK9, and to the BU 4061T development of powerful animal genetic models.
inhibitory small molecules, which are not yet available. Annexin A2 is strongly expressed in lungs, aorta, heart, adrenals and small intestine. Intracellular AnxA2 is part of a heterotetramer complex comprising two AnxA2 monomers and two copies of its natural binding partner, p11. AnxA2 is composed of an N-terminal 23 aa segment that binds p11, followed by four repeat structures. Although lacking a signal peptide, AnxA2 is translocated to the cell surface through a p11-dependent mechanism and is found at the cell surface of epithelial and endothelial cells. As a co-receptor for plasminogen and tissue plasminogen activator, which are required for plasmin generation, the AnxA2 complex promotes vascular fibrinolysis. It was suggested that injection of AnxA2 in human may improve thrombotic disease outcome. AnxA2 knockout mice are viable, but deficient in endothelial plasminogen processing into plasmin and neoangiogenesis. We previously identified AnxA2 as a natural inhibitor of PCSK9’s function on the LDLR, through binding of the first R1 repeat domain of AnxA2 to the CHRD. Inhibition likely occurs via an allosteric PCSK9 conformational change induced by AnxA2, similar to what was reported for mAbs that bind the CHRD. In this report, we investigated in more details the molecular interaction of the R1 domain of AnxA2 and PCSK9 and showed that a synthetic peptide spanning the R1 domain is a potent inhibitor. We further hypothesized that PCSK9 is much more active in enhancing LDLR degradation in AnxA22/2 mice. Indeed, our data showed that AnxA22/2 mice exhibit a higher level of MLN4924 circulating PCSK9 and lower LDLR protein levels in various tissues previously reported to be refractory to PCSK9’s extracellular function on LDLR, such as the adrenals. Furthermore, overexpression of AnxA2 using a recombinant adenovirus resulted in higher LDLR levels in liver. Finally, by sequencing exons of human AnxA2 we identified individuals with a V98L polymorphic variation in the R1 domain, and showed that this variant could be associated with lower levels of circulating PCSK9. The discovery of PCSK9 and its genetic relation to hypercholesterolemia led to a very exciting and active period of identification of the mechanisms of action of PCSK9, and to the BU 4061T development of powerful animal genetic models.
This has reached maturity to the point that we can entertain various viable therapeutic options
Leave a reply